

Original
Regiones codificantes con baja cobertura en la secuenciación masiva de genes de hipoacusia no sindrómica autosómica dominante
Encoding regions with low coverage in massive sequencing of genes of no syndromic autosomal dominant hearing loss
Actual. Med. 2017; 102: (802): 135-139 DOI: 10.15568/am.2017.802.or01
Enviado: 31-10-2017
Revisado: 12-11-2017
Aceptado: 06-12-2017
RESUMEN
Introducción: La hipoacusia es una de las alteraciones sensoriales más frecuentes, afectando aproximadamente a uno de cada 500 nacimientos, y pudiendo producirse a cualquier edad en individuos sanos. Gracias a las técnicas de secuenciación masiva, como el análisis del exoma completo, y al uso de estrictos protocolos de filtrado de variantes, se han identificado más de 100 loci relacionados con la hipoacusia no sindrómica. El objetivo de este estudio es diseñar un protocolo para la identificación de las secuencias nucleotídicas de baja cobertura a partir de datos de secuenciación de exoma, particularmente en los genes causales de hipoacusia no sindrómica autosómica dominante.
Materiales y métodos: Cincuenta y cinco individuos (15 pacientes con enfermedad de Meniere y 40 controles) fueron seleccionados para llevar a cabo un análisis genómico mediante secuenciación de exoma completo. Los exones de los genes asociados a hipoacusia no sindrómica autosómica dominante de estas 55 muestras fueron analizados mediante el software Integrative Genome Viewer. Los datos de cobertura se contrastaron con las bases de datos de secuencias de exoma y genoma actuales. Finalmente, se simularon las estructuras primaria y secundaria de las secuencias implicadas con la herramienta bioinformática mfold.
Resultados: Trece de los 34 genes de hipoacusia no sindrómica autosómica dominante presentaron una baja cobertura. Veinte de las 24 secuencias exónicas con baja cobertura presentaban un contenido de GC más elevado del considerado como óptimo, y todas ellas contenían, al menos, una estructura secundaria. Los genes con un mayor número de exones con baja cobertura son MYH14, WFS1 y P2RX2.
Conclusiones:
- La secuenciación del exoma humano muestra que muchos de los genes de hipoacusia autosómica no sindrómica dominante presentan baja cobertura en algunos de sus exones.
- El análisis del contenido de guaninas y citosinas, y la presencia de estructuras secundarias determinan secuencias específicas con problemas de lectura.
- Es necesario validar los resultados obtenidos por la Secuenciación de Nueva Generación para evitar posibles falsos positivos.
Palabras clave: hipoacusia, genética, exoma, bioinformática.
ABSTRACT
Introduction: Hearing loss is one of the most frequent sensory alterations, affecting approximately one in 500 births, and it may occur de novo at any age in healthy individuals. Thanks to massive sequencing techniques, such as whole exome sequencing, and the use of strict variant filtering protocols, more than 100 loci related to non-syndromic hearing loss have been identified. The aim of this study is to design a protocol for the identification of low coverage nucleotide sequences from exome sequencing data, particularly in the genes responsible for non-syndromic autosomal dominant hearing loss.
Materials y methods: Fifty-five individuals (15 cases with Meniere’s disease and 40 controls) were selected to perform a genomic analysis by whole exome sequencing. The exons of the genes related to non-syndromic autosomal dominant hearing loss of the whole sample were analyzed with Integrative Genome Viewer. The coverage was evaluated with the current genomic and exomic databases. Finally, we simulated the primary and secondary structures of the sequences involved with the mfold bioinformatic tool.Results: Thirteen from the 34 genes involved in no syndromic autosomal dominant hearing loss had low coverage. Twenty from the 24 exonic sequences with low coverage had higher percentage of GC content than the optimal and all of them had one or more secondary structures. Genes with more exons with low coverage were: MYH14, WFS1 and P2RX2.
Conclusions:
- Whole Exome Sequencing shows that most of the non-syndromic autosomal dominant hearing loss genes had low coverage in some exons.
- The analysis of the guanines and cytosines content, and the presence of secondary structures have determined specific sequences with read problems.
- The results of Next Generation Sequencing must be validated to avoid possible false positives.
Keywords: hearing loss, genetics, exome, bioinformatics.
Leer Artículo Completo
INTRODUCCIÓN
La hipoacusia es una de las alteraciones sensoriales más frecuentes, afectando aproximadamente a uno de cada 500 nacimientos, y pudiendo producirse de novo a cualquier edad en individuos sanos (1). Su etiología es diversa, pudiendo originarse tanto por factores medioambientales (ruido, virus o sustancias ototóxicas) como por factores genéticos. A diferencia de los factores medioambientales, que pueden ser reducidos o prevenidos a través de medidas de concienciación pública, la hipoacusia generada por un trasfondo genético sigue presentando grandes retos. Clínicamente, la hipoacusia se puede clasificar como sindrómica o no sindrómica dependiendo, respectivamente, de si se presenta junto a otro fenotipo clínico (discapacidad visual, enfermedad renal crónica…) o no. Las hipoacusias no sindrómicas son las más frecuentes, presentándose en, aproximadamente, el 70% de los casos (2,3).
Gracias a las técnicas de secuenciación masiva, como el análisis del exoma completo (WES), y al uso de estrictos protocolos de filtrado de variantes, se han identificado más de 100 loci relacionados con la hipoacusia no sindrómica (4-6). La identificación de variantes genéticas que causan el deterioro de la audición ha permitido un mejor conocimiento de las vías moleculares que están involucradas en la regulación de la percepción auditiva. El conocimiento de estas vías nos proporciona un punto de partida para el desarrollo de opciones terapéuticas para las personas afectadas por enfermedades relacionadas con la audición (7). La eficacia del WES y el filtrado de variantes es muy alta debido a que el 85% de las variantes causantes de enfermedades se encuentran en regiones exónicas (8). Además, el exoma solo representa 1-2% del genoma, lo que reduce considerablemente el coste económico y facilita el tratamiento de los datos (9). Como contrapartida, la cobertura del WES es del 90%, existiendo de esta manera regiones codificantes de difícil lectura de las que no se obtienen suficientes datos. Esto se debe a problemas inherentes a las estructuras secundarias que pueden adoptar los exones o a su contenido de bases GC. Las estructuras de difícil lectura pueden deberse a repeticiones (ACC, CCG…), repeticiones Alu, regiones horquilla o bucles, colas poli-A/T y otros motivos que causan compresión en el ADN. Por otro lado, el contenido de GC mínimo que causa problemas aún no está claramente determinado, pero se sitúa en torno al 60-65% en regiones de al menos 100 pares de bases (10). Todo ello conlleva una enorme dificultad para secuenciar ciertos fragmentos de ADN y da lugar a regiones en las que se ha perdido total o parcialmente la información. Por consiguiente, esto implica reanalizar estas regiones con otras técnicas más precisas pero más costosas, como es el re-análisis del genoma completo o la secuenciación Sanger. La identificación de estas regiones in sílico puede prevenir la pérdida de regiones del genoma relevantes y que podrían incluir variantes candidatas. Por tanto, el desarrollo de un protocolo de identificación de estas regiones es un paso crucial en la identificación variantes en las enfermedades poco frecuentes que puede concluir en investigaciones de alto impacto y una mejor comprensión de las bases biológicas de las enfermedades hereditarias (11).
El objetivo de este estudio es diseñar un protocolo para la identificación de las secuencias nucleotídicas de baja cobertura (secuencias de muy difícil lectura) a partir de datos de secuenciación de exoma, particularmente en los genes dominantes relacionados con hipoacusia no sindrómica.
MATERIALES Y MÉTODOS
Pacientes y secuenciación del exoma completo
Cincuenta y cinco individuos (15 casos con enfermedad de Meniere y 40 controles), fueron seleccionados para obtener muestras de sangre, realizar la extracción de ADN y la secuenciación del exoma completo (12-14). El ADN fue aislado a partir de sangre periférica mediante QIAamp DNA Mini Kit (Qiagen). Los exones y regiones circundantes (UTR 3’ y 5’) fueron capturadas mediante el kit Agilent’s All Exon 50MB. Las condiciones y los cebadores empleados se encuentran disponibles en la casa comercial. Las muestras fueron secuenciadas en la plataforma SOLiD5500xl. El alineamiento y el mapeo se realizó mediante el uso del software Lifescope v2.5 y el genoma de referencia GRCh37/hg19, dando lugar a un archivo .BAM por individuo.
Este estudio con muestras biológicas humanas fue aprobado por el Comité de Ética de Investigación del Hospital y se siguieron los estándares éticos reconocidos por la Ley Biomédica de España (15) y los principios de la Declaración de Helsinki de 1975 (16). Todos los pacientes participantes en el estudio fueron informados de los objetivos del mismo y firmaron el consentimiento informado.
Análisis bioinformático
- Análisis de exones en genes dominantes asociados a hipoacusia no sindrómica.
Los archivos .BAM fueron visualizados en el software Integrative Genome Viewer (IGV) para detectar fallos de secuenciación en los exones de los 34 genes de hipoacusia no sindrómica autosómica dominante descritos en la base de datos de hipoacusia hereditaria (http://hereditaryhearingloss.org/). Los exones fueron clasificados en 3 categorías según la calidad de su lectura: “Buena” para aquellos con una cobertura >20 lecturas (>20X); “Parcial” para una cobertura entre 5-20 lecturas (5-20X) y “Mala” para una cobertura <5 (<5X). Todos los exones que presentaban fallos de lectura en el 100% de las muestras fueron comparados frente a las bases de datos ExAC y GnomAD (17). Los exones fueron numerados, siendo siempre el primero el de la izquierda obviando la dirección de la lectura del gen. Las secuencias que comprendían estos exones fueron analizadas mediante un script realizado ad hoc por el grupo de Otología y Otoneurología para estimar el número de bases que contenían. Para la búsqueda de estructuras secundarias en estas secuencias fue usada la herramienta virtual mfold (18). Estas estructuras fueron generadas bajo las condiciones presentes durante la secuenciación en la plataforma de secuenciación SOLiD 5500xl: 98ºC de temperatura y 3M de concentración de sodio.
RESULTADOS
Genes dominantes causales de hipoacusia autosómica no sindrómica
En conjunto, los 34 genes de hipoacusia no sindrómica autosómica dominante estudiados comprenden un total de 592 exones. Veinticuatro de estos exones, pertenecientes a 13 genes, presentan una baja cobertura. Los exones fueron clasificados como “Ilegibles” cuando un exón presentó baja cobertura en el 100% de las muestras (Tabla 1-2). Los resultados muestran que genes muy destacados para la hipoacusia (MYH14, WFS1 y P2RX2) presentan mala cobertura en muchos de sus exones. Además, 20 de las 24 secuencias exónicas presentan un contenido de GC más elevado del considerado como óptimo (Tabla 2). Las secuencias que alcanzaron los valores más elevados de GC fueron: el exón 1 de KCNQ4 con un 79,55%; el exón 2 de COCH con un 74.47%; el exón 1 de MYH14 con un 72,77%; el exón 1 de P2RX2 con un 72,09% y el exón 27 de DIAPH1 con un 71.55%. Por otro lado, cabe destacar que el exón 9 de HOMER2 tiene 5 pares de bases, siendo 3 de las bases citosinas, lo que posiblemente causa la dificultad de su lectura. Finalmente, se identificó que el exón 28 de DIAPH1 y el exón 1 de MYH14 presentan estructuras secundarias especialmente complejas respecto a las demás. El exón 1 de KCNQ4 fue el que presentó más bucles en su secuencia (Figura 1).
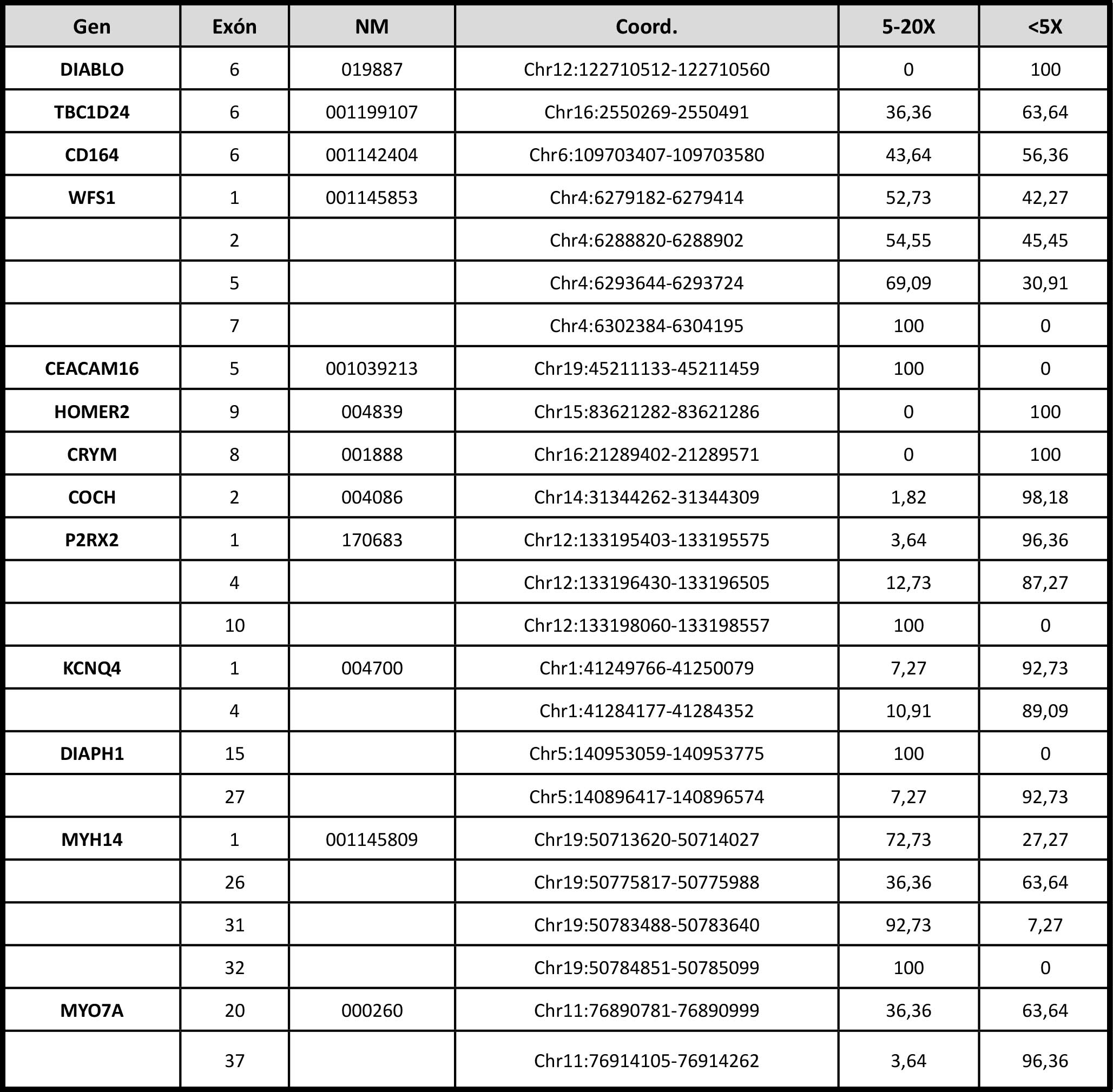
Tabla 1. Genes de hipoacusia no sindrómica autosómica dominante con exones “Ilegibles”. Exones con baja cobertura en un 100% de las muestras (sumando 5-20X con <5X). NM: Transcrito NM. Coord.: Coordenadas cromosómicas. 5-20X: porcentaje de muestras con un número de lecturas de entre 5-20. <5X: porcentaje de muestras con un número lecturas por debajo de 5.
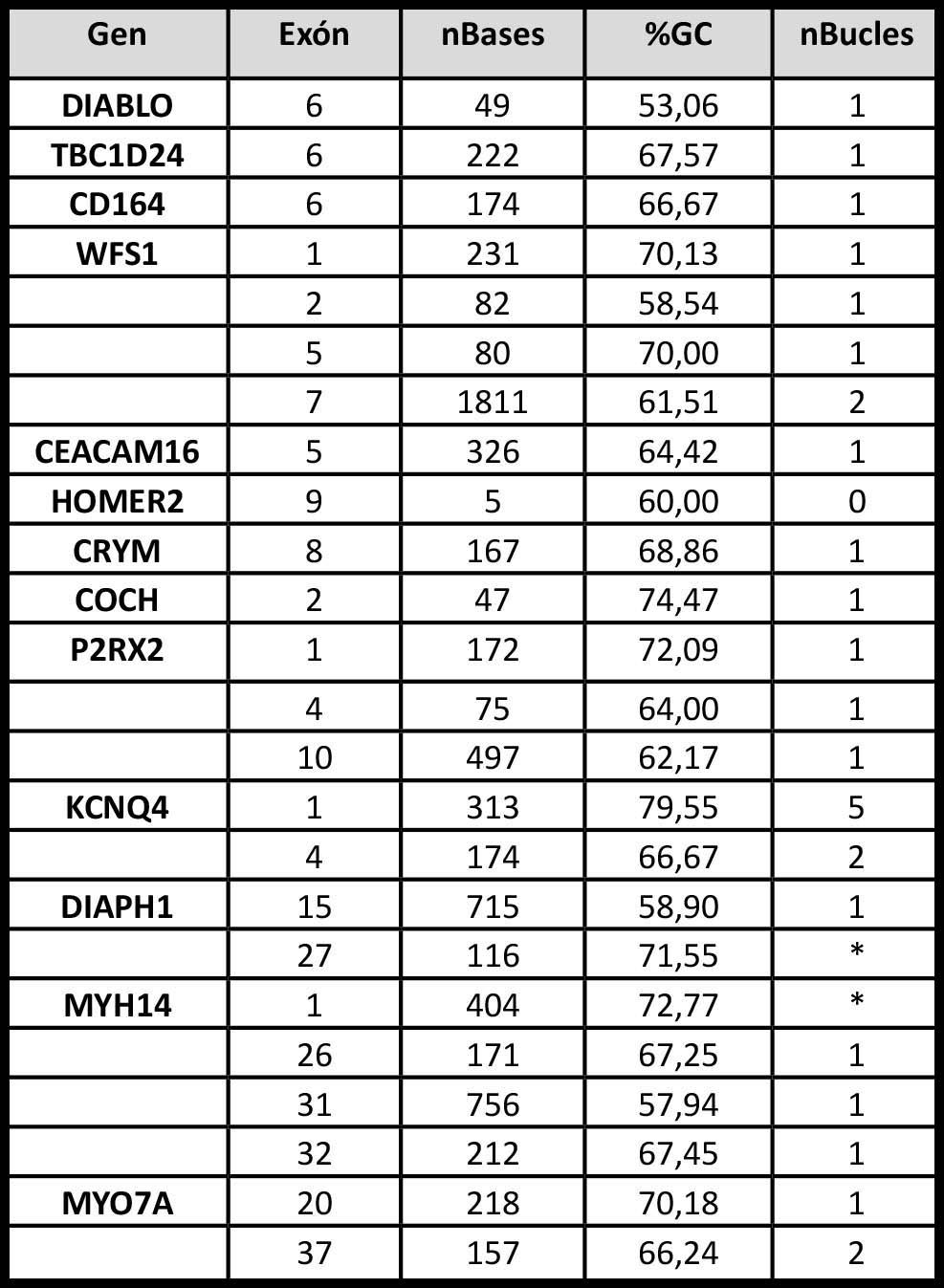
Tabla 2. Estructura primaria y secundaria de los genes de hipoacusia no sindrómica dominante con exones “Ilegibles”. nBases: número de bases totales de la secuencia. %GC: porcentaje de bases GC de la secuencia. nBucles: número de estructuras secundarias encontradas en la secuencia. *: Estructura compleja.
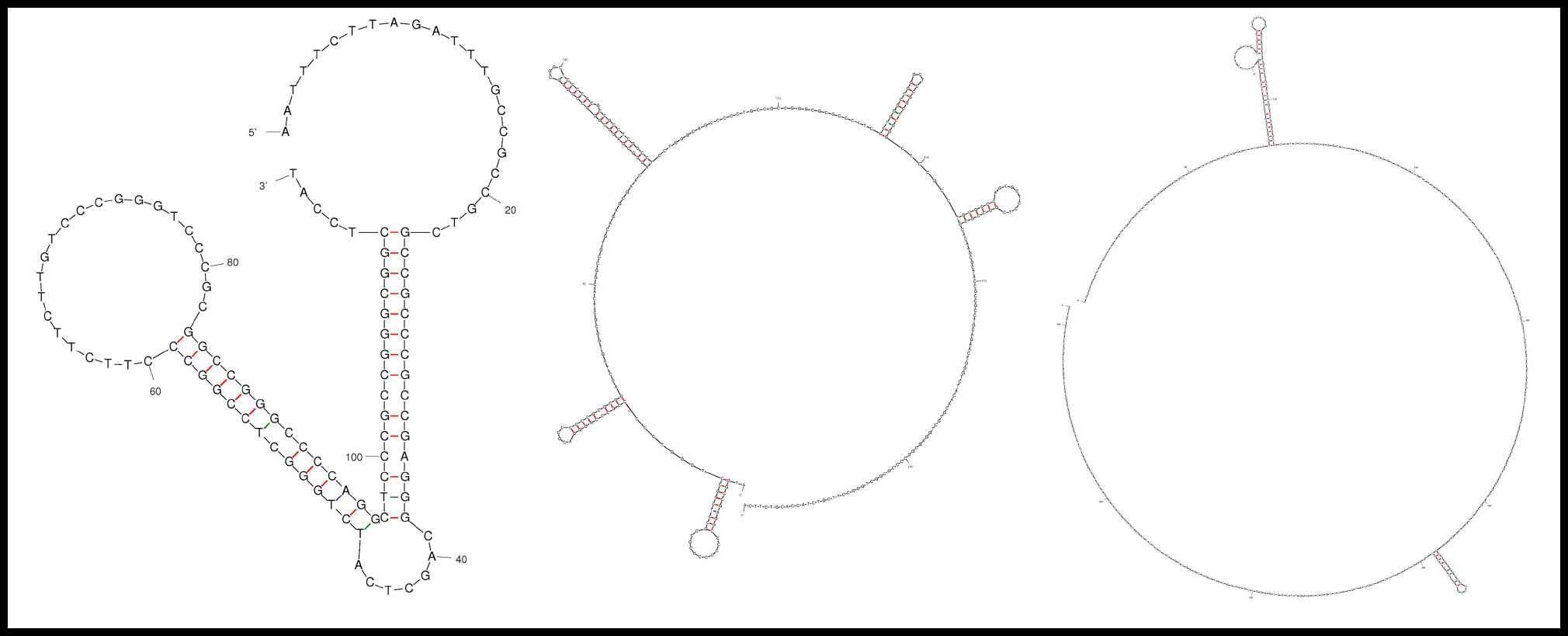
Figura 1. Estructuras secundarias destacables en algunos genes de hipoacusia no sindrómica autosómica dominante. Las secuencias fueron seleccionadas por el número de estructuras secundarias o la complejidad de estas. A: Exón 27 de DIAPH1. B: Exón 1 de KCNQ4. C: Exón 1 de MYH14.
DISCUSIÓN
Las tecnologías de secuenciación masiva del genoma tienen algunas limitaciones que estamos comenzando a descubrir. Existen regiones que, bien por su estructura primaria con alto contenido en GC o por su estructura secundaria, son difíciles de secuenciar. Estas regiones pueden contener variantes causales de enfermedades poco frecuentes, como la hipoacusia neurosensorial o la enfermedad de Meniere. Según los resultados de este estudio, algunos de los genes dominantes relacionados con hipoacusia tienen un número considerable de exones con baja cobertura (MYH14, WFS1 y P2RX2). En primer lugar, MYH14 está descrito como un gen de hipoacusia importante, el cual causa hipoacusia no sindrómica autosómica dominante prelocutiva grave. Se han descrito dos variantes no sinónimas en este gen (c.572A>G y c.73C>T) mediante secuenciación dirigida, un método de Secuenciación de Nueva Generación (NGS). La segunda variante se encuentra en el exón 2 (19), una región que en nuestros datos de secuenciación muestran baja cobertura en el 100% de las muestras y que, además, bajo las condiciones de la plataforma SOLiD, forma una estructura secundaria compleja. Por otro lado, variantes en el gen WFS1 están directamente relacionadas con hipoacusia no sindrómica de baja frecuencia. Se han identificado 2 mutaciones que provocan la aparición de un codón de STOP prematuro (codón sinsentido) en el exón 8 deeste gen (c.2086C>T y c.2108G>A) mediante análisis de ligamiento (20). Nuestro estudio muestra que este exón tiene baja cobertura en el 100% de las muestras y presenta un contenido de GC problemático según la bibliografía (61.51%). Por último, el gen P2RX2 está involucrado en el correcto funcionamiento de los canales iónicos P2X2 activados por ATP que se encuentran en las células sensoriales y de soporte de la cóclea. Mediante un análisis de ligamiento, Faletra et al. han descrito una mutación sinsentido (c.1057C>G) que afecta a un residuo altamente conservado entre las especies (21). Esta mutación ocurre en el exón 10, el cual presenta baja cobertura en el 100% de las muestras de nuestro estudio. Además, esta secuencia presenta un contenido alto de GC (62.17%).
DIAPH1 y KCNQ4 son otros de los genes destacados en nuestro estudio. Por un lado, el gen DIAPH1 codifica una proteína encargada de unirse a los extremos de actinas sin ramificar para reducir la velocidad de despolimerización y polimerización de estas regiones. La variante c.3634+1G>T causa DFNA1, enfermedad caracterizada por una hipoacusia progresiva que comienza desde la infancia. Esta variante ocurre cerca del extremo C-terminal, el cual interacciona con el N-terminal, produciendo la activación de DIA1, proteína que juega un papel importante en la morfología celular y la organización del citoesqueleto. Por otro lado, se ha descrito una variante (c.3610C>T) en el exón 27 que resulta en un incremento de la polimerización de actina, produciéndose microvellosidades alargadas (22). El exón próximo a esta región C-terminal (exón 27) tiene baja cobertura en el 100% de las muestras, consta de un elevado contenido de GC (71.55%) y la estructura que se puede originar durante la secuenciación es especialmente compleja.
Por otro lado, el gen KCNQ4 codifica un canal iónico de potasio que juega un papel fundamental en la fisiología de esta enfermedad autosómica dominante progresiva relacionada con hipoacusia de alta frecuencia. Kamada et al. (23) identificaron una deleción heterocigótica de 1 pb (211delC) en el exón 1 de KCNQ4 que daba lugar a un truncamiento de la proteína, perdiéndose los dominios transmembrana. Los individuos afectados por esta mutación presentaban síntomas mucho menos severos que otros pacientes con mutaciones sinsentido en KCNQ4. Nuestro estudio ha determinado que el exón 1 de KCNQ4 tiene baja cobertura en el 100% de las muestras, así como un alto contenido de GC (79.55%) y, de todos los exones afectados, es el que más estructuras secundarias presentaba en su secuencia.
Veinte de los 24 exones de baja cobertura identificados presentan un elevado contenido de GC (>60%). Estudios que respaldan este intervalo nos muestran que el uso de tampones, aditivos (DMSO, betaína), métodos de purificación, el medio de carga y las condiciones de los ciclos no juegan un papel determinante en la calidad de la secuenciación estadísticamente significativo. Sin embargo, el uso de cebadores pequeños (<3 pmol) si produce una secuenciación con menos cobertura y peor calidad (24). Por otro lado, la presencia de estructuras secundarias de tipo horquilla, como las que observamos en los exones de DIAPH1 y KCNQ4,dificultan la correcta secuenciación de estas regiones, tal y como se nos muestra en estudios realizados con plásmidos (25). Todos los exones presentaron, al menos, una horquilla en su secuencia. No obstante, la presencia de las llamadas colas de poli-A o poli-T y otros elementos, como continuas repeticiones y motivos 5’-YGN1–2AR (Y=primidina, R=purina, N=cualquier base), también afectan a la calidad de la secuenciación (24, 25). Estos resultados se deben a la baja sensibilidad de la técnica (~73%) que puede resultar en inespecificidades de los cebadores. Estos datos hacen ver que, independientemente del método usado, todos los resultados de la secuenciación masiva deben ser inmediatamente validados mediante secuenciación Sanger, especialmente las variantes candidatas (26). Solo de este modo podremos afirmar que una variante no es un falso positivo.
A medida que este tipo de problemas vayan desapareciendo en las técnicas de secuenciación e identificación de variantes, el análisis genético de variantes comunes será más usado en algunos entornos clínicos. El precio cada vez más bajo de las técnicas de secuenciación junto al mayor conocimiento sobre las variantes comunes que predisponen a hipoacusia hacen pensar que las pruebas genéticas irán ganando espacio en el diagnóstico de la hipoacusia, permitiendo un diagnóstico precoz que permita a los pacientes proteger su audición.
CONCLUSIONES
- Los datos de secuenciación del exoma humano muestran que la mayoría de los genes de hipoacusia autosómica dominante presentan baja cobertura en al menos un exón.
- El análisis del contenido de GC y la presencia de estructuras secundarias han determinado secuencias específicas con problemas de lectura.
- Es necesario validar los resultados obtenidos por la Secuenciación de Nueva Generación para evitar posibles falsos positivos.
REFERENCIAS
- Thompson DC, McPhillips H, Davis RL, Lieu TL, Homer CJ, Helfand M. Universal newborn hearing screening: summary of evidence. JAMA. 2001; 286(16):2000-10.
- Shearer AE, Hildebrand MS, Smith RJH. Hereditary hearing loss and deafness overview. GeneReviews; 1999-2017.
- Morton CC, Nance WE. Newborn hearing screening–a silent revolution. N Engl J Med. 2016; 354(20):2151-64.
- Angeli S, Lin X, Liu XZ. Genetics of hearing and deafness. Anat Rec. 2012; 295, 1812–1829
- Richardson GP, de Monvel JB, Petit C. How the genetics of deafness illuminates auditory physiology. Annu Rev Physiol. 2011; 73, 311–334.
- Dror AA, Avraham KB. Hearing impairment: a panoply of genes and functions. Neuron. 2010; 68, 293–308
- Ulrich M, Barr-Gillespie PG. New treatment options for hearing loss. Nature Rev Drug Disc. 2015.
- Rabbani B, Tekin M, Mahdieh N. The promise of whole-exome sequencing in medical genetics. J Hum Genet. 2014; 59(1):5–15.
- Warr A, Robert C, Hume D, Archibald A, Deeb N, Watson M. Exome Sequencing: Current and Future Perspectives. G3 (Bethesda). 2015; 5(8):1543–50.
- Kieleczawa J. Fundamentals of sequencing of difficult templates–an overview. J Biomol Tech. 2006; 17(3):207–17.
- López-Escamez JA. Papel de la medicina genómica en las enfermedades del oído medio e interno. Acta Otorrinolaringol. 2012; 63(6):470-479.
- Requena T, Cabrera S, Martin-Sierra C, Price SD, Lysakowski A, Lopez-Escamez JA. Identification of two novel mutations in FAM136A and DTNA genes in autosomal-dominant familial Meniere’s disease. Hum Mol Genet. 2015; 24(4):1119–26.
- Martín-Sierra C, Requena T, Frejo L, Price SD, et al. A novel missense variant in PRKCB segregates low-frequency hearing loss in an autosomal dominant family with Meniere’s disease. Hum Mol Genet. 2016; 25(16):3407–15.
- Martín-Sierra C, Gallego-Martinez A, Requena T, Frejo L, Batuecas-Caletrío A, Lopez-Escamez JA. Variable expressivity and genetic heterogeneity involving DPT and SEMA3D genes in autosomal dominant familial Meniere’s disease. Eur J Hum Genet. 2017; 25(2):200–7.
- España. Ley 14/2007, de 3 de Julio, de Investigación Biomédica. Boletín Oficial del Estado; 4 de Julio de 2007. pp. 28826-28848.
- World Medical Association. World Medical Association Declaration of Helsinki. JAMA. 2013 Nov 27; 310(20):2191.
- Lek M, Karczewski KJ, Minikel E V, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536(7616):285–91.
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003; 31(13):3406–15.
- Kim BJ, Kim AR, Han JH, Lee C, Oh DY, Choi BY. Discovery of MYH14 as an important and unique deafness gene causing prelingually severe autosomal dominant non-syndromic hearing loss. J Gene Med. 2017; 19(4):e2950.
- Sun Y, Cheng J, Lu Y, et al. Identification of two novel missense WFS1 mutations, H696Y and R703H, in patients with non-syndromic low-frequency sensorineural hearing loss. J Genet Genomics. 2011; 38(2):71–6.
- Faletra F, Girotto G, D’Adamo AP, Vozzi D, Morgan A, Gasparini P. A novel P2RX2 mutation in an Italian family affected by autosomal dominant non-syndromic hearing loss. Gene. 2014; 534(2):236–9.
- Ueyama T, Ninoyu Y, Nishio S, et al. Constitutive activation of DIA1 (DIAPH1) via C-terminal truncation causes human sensorineural hearing loss. EMBO Mol Med. 2016; 8(11):1310–24.
- Kamada F, Kure S, Kudo T, et al. A novel KCNQ4 one-base deletion in a large pedigree with hearing loss: implication for the genotype-phenotype correlation. J Hum Genet. 2006; 51(5):455–60.
- Kieleczawa J. Fundamentals of sequencing of difficult templates–an overview. J Biomol Tech. 2006; 17(3):207–17.
- Yamakawa H, Nakajima D, Ohara O. Identification of sequence motifs causing band compressions on human cDNA sequencing. DNA Res. 1996; 3(2):81–6.
- Chang Y-S, Huang H-D, Yeh K-T, Chang J-G. Evaluation of whole exome sequencing by targeted gene sequencing and Sanger sequencing. Clin Chim Acta. 2017 Aug; 471:222-32.
INFORMACIÓN DEL ARTÍCULO
Agradecimientos: A José Antonio López Escamez, por la revisión crítica de este manuscrito.
Este Trabajo forma parte del Trabajo fin de Master de Ingeniería Titular y Terapias Avanzadas de Iván Morales Esquina.
Conflicto de intereses: Los autores declaran no tener ningún conflicto de intereses.
Autor para la correspondencia: Teresa Requena. Otology & Neurotology Group CTS495, GENYO, -Centre for Genomics and Oncological Research- Pfi zer/University of Granada/Andalusian Regional Government. Avda de la Ilustración, 114 · 18016 Granada SPAIN. Phone. +34 958 715 500-160. E-mail: mariateresa.requena@genyo.es





