

Caso Clínico
Linfoma maligno enmascarado como mieloma múltiple
Malignant lymphoma masquerading as multiple myeloma
Actual. Med. 2017; 102: (802): 173-175 DOI: 10.15568/am.2017.802.cc02
Enviado: 01-12-2017
Revisado: 06-12-2017
Aceptado: 15-12-2017
RESUMEN
Los hallazgos de hipercalcemia, lesiones líticas craneales, anemia de trastornos crónicos y lesión renal aguda, suelen ser característicos del mieloma múltiple. En el presente documento se describe un caso interesante de linfoma anaplásico de células grandes con características que imitaban un mieloma múltiple.
Palabras clave: Linfoma maligno, Mieloma múltiple, Hipercalcemia, Anemia, Lesión renal aguda, Lesiones osteolíticas
ABSTRACT
The findings of hypercalcemia, cranial lytic lesions, anemia of chronic disorders and acute renal injury are often characteristic of multiple myeloma. Here we describe an interesting case of large cell anaplastic lymphoma with features mimicking multiple myeloma.
Keywords: Malignant lymphoma, Multiple myeloma, Hypercalcemia, Anemia, Acute kidney injury, Bone lytic lesions
Leer Artículo Completo
INTRODUCCIÓN
El mieloma múltiple se caracteriza por una proliferación descontrolada de células plasmáticas monoclonales en la medula ósea, lo que lleva a una producción anómala de inmunoglobulinas y de sus cadenas.
El mieloma múltiple es responsable de 1 % de todo el cáncer a nivel mundial y de 10 a 15 % de las neoplasias hematológicas. El riesgo de desarrollar mieloma múltiple es mayor en grupos de edad avanzada. El promedio de edad de aparición es de 71 años en hombres y de 74 años en mujeres1.
En la mayoría de los pacientes la enfermedad se desarrolla a partir de la gamapatía monoclonal de significado incierto (MGUS) la cual se diagnostica comúnmente de manera incidental; MGUS confiere un riesgo de 1% por año de progresión a mieloma múltiple. Otra fase transicional es el mieloma latente la cual confiere un riesgo de progresión de 10% por año. Ambas entidades, a diferencia del mieloma múltiple sintomático, se caracterizan por ausencia de daño a órgano blanco2.
El diagnóstico definitivo de mieloma múltiple se hace utilizando los criterios del International Myeloma Working Group los cuales son:
- Mayor de 10% de células plasmáticas clonales en médula ósea o biopsia que evidencie plasmocitoma óseo o extramedular.
Y cualquiera de los siguientes eventos definitorios de mieloma: - Evidencia de daño a órgano blanco (CRAB): hipercalcemia >11 mg/dL, insuficiencia renal GFR <40 ml/min, anemia >2 g/dL por abajo del límite inferior y lesiones osteolíticas.
- Cualquiera de los siguientes biomarcadores de malignidad: Más de 60% de células plasmáticas monoclonales en médula ósea, razón de cadenas ligeras libres en suero involucradas:no-involucradas >100, más de una lesión focal por resonancia magnética3.
Existen cambios citogenéticos que se asocian a un mal pronóstico los cuales son evidenciados por hibridación fluorescente in situ (FISH). Estos son: t(4;14) y deleción 17p13 4.
Los linfomas se originan del tejido linfoide. Se desarrollan como consecuencia de la expansión clonal de una u otra línea linfoide (linfocitos B o T y más raro NK), dando los dos grandes grupos: Linfoma Hodgkin (LH) y linfoma no Hodgkin. El linfoma no Hodgkin (LNH) comprende un grupo de enfermedades relacionadas entre sí́. Cada variedad histológica de linfoma no Hodgkin se caracteriza por la transformación maligna de las células linfoides, con morfología, inmunofenotipo, genética y clínica diferente. El linfoma anaplásico de células grandes (LACG) es un grupo heterogéneo de linfoma no Hodgkin, descrito por Stein y col. en 19855.Histológicamente está compuesto por células linfoides grandes anaplásicas con patrón de crecimiento cohesivo con propensión a invadir sinusoides de los ganglios linfáticos y exhibe expresión constante de CD30 (linfoma Ki-1)6. Los LACG están compuestos por células T o células nulas (Null) y se presentan más frecuentemente en las primeras tres décadas de la vida, tienen diferencias clínicas y moleculares con las neoplasias histológicamente similares que presentan inmunofenotipo B. Por lo anterior, la Organización Mundial de la Salud (OMS) ha propuesto que el término de LACG sea restringido a los linfomas con in- munofenotipo T y nulo. Algunos LACG presentan la translocación t(2;5) (p23;q35) que resulta en la producción de la proteína ALK-1, que se manifiesta más en pacientes jóvenes y les confiere mejor pronóstico7.
La estatificación se realiza con la escala de Ann Arbor la cual toma en cuenta la afectación ganglionar tanto en número como en distribución supra e infra diafragmática, si hay compromiso extraganglionar y si existen síntomas B 8.
Existe en la literatura un reporte de caso en donde se describe un linfoma no Hodgkin enmascarado por datos clínicos de mieloma múltiple. Se trata de un paciente masculino de 67 años originario de Hong Kong el cual tenía antecedente de dolor óseo generalizado, pérdida de 6 kg en tres semanas así como debilidad y poliuria de una semana de evolución. Una presentación clínica típica de mieloma múltiple. A la exploración física solo presentaba datos de deshidratación y adenopatías cervicales. Debido a la edad del paciente y el dolor óseo haría pensar en mieloma múltiple pero después de realizar biopsia ganglionar se hizo el diagnóstico de linfoma 9.
La coexistencia de HL y MM es rara pero existe un reporte de caso que describe a un paciente masculino de 45 años originario de China el cual fue diagnosticado de HL estadio III, durante su abordaje diagnostico se le realizó TAC y RMI las cuales mostraron lesiones osteolíticas, aumento de las cadenas IgA λ, y células plasmáticas aumentadas en el aspirado de médula ósea por lo que también se le diagnosticó mieloma múltiple 10.
CASO CLÍNICO
Masculino de 74 años con antecedentes de etilismo y tabaquismo crónicos, con cuadro clínico de 8 meses de evolución caracterizado por por pérdida de peso no intencioanda de 20 kilogramos, acompañado de anorexia, diaforesis nocturna y fiebre vespertina no cuantificada. A su ingreso a la unidad de urgencias se encontró con signos vitales dentro de los parámetros normales, con laboratorios que integraban una anemia normocítica normocrómica grado II OMS (Hemoglobina 8.8 g/dL) se solicitó cinética de hierro, vitamina B12 y folatos como parte de su abordaje, resultando un síndrome anémico por trastornos crónicos , Lesión renal aguda (Creatinina sérica 1.5 mg/dL), e Hipercalcemia (Calcio corregido 14.6 mg/dL) sin repercusión electrocardiográfica, con Antígeno prostático específico 3.49 ng/dL. A la exploración física se observó caquéxico, con palidez mucotegumentaria y tacto rectal sin alteraciones. Se realizó serie ósea radiológica, que reveló radiografía de cráneo con presencia de lesiones líticas en sacabocado en hueso frontal en patrón de “sal y pimienta” (Figura 1).
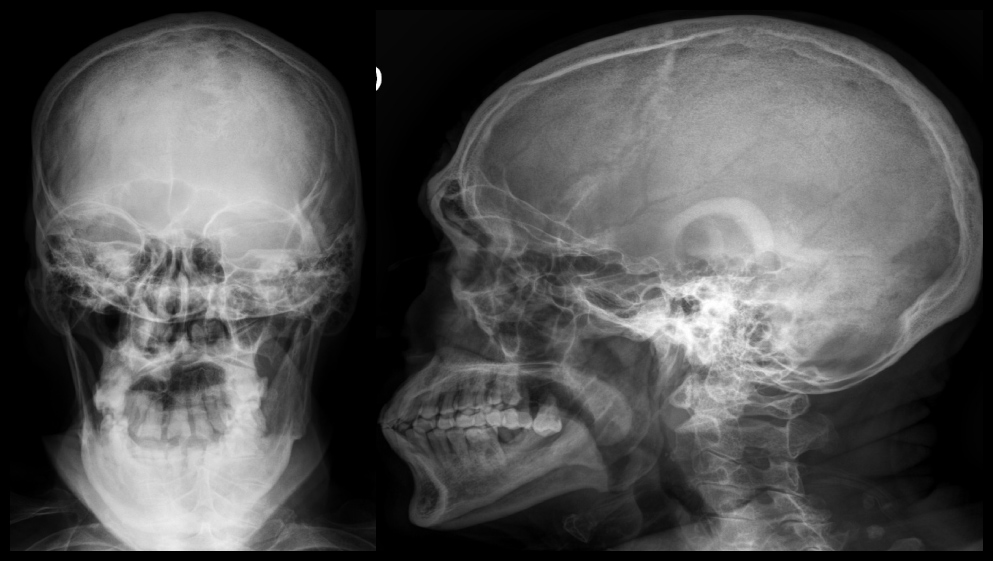
Figura 1. Radiografía de cráneo: Presencia de lesiones líticas en sacabocado en hueso frontal en patrón de “sal y pimienta”.
Se realizó prueba de función hepática con Proteínas Totales en 6.71 g/dL y Globulina 4.39 g/dL, Inmunoglobulinas, unicamente con IgG ligeramente elevada 1098 mg/dL, resto normal, PTH (Parathormona) suprimida y Péptido relacionada con la PTH (PTHrP) negativo. Por la alta sospecha de mieloma múltiple, se realizó electroforesis de proteínas en suero que demostró: ALFA1 5.4% (2-3.5 %), ALFA1 0.26 (0.2-0.3 g/dL), ALFA2 0.84 g/dL (0.4-0.8 g/dL), ALFA2 17.7% (5.4-10.6%), GAMMA 1.23 g/dL (0.6-1.3 g/dL), GAMMA 25.7% (8-18%), resto dentro de la normalidad al igual que la electroforesis en orina. Posteriormente se realizó TAC de abdomen con evidencia de conglomerado ganglionar retroperitoneal y biopsia y aspirado de médula ósea con infiltración de céulas malignas, 8% de células plasmáticas de distribución perivascular e intersticial, observandose médula ósea fibrosa con céulas grandes de abundante citoplasma, núcleos grandes y nucléolo prominente (Figura 2), por tanto se realizó inmunohistoquímica y biopsia de ganglios retroperitoneales que evidenciarón linfoma anaplásico de céulas grandes, en donde las células neoplásicas fueron positivas para CD45 y a los marcadores T: CD45RO, CD43, CD3, CD4 y CD8, expresando en membrana y a nivel paranuclear EMA y CD30 como positividad al ALK-1 en citoplasma y núcleos (Figura 3). El paciente fue tratado con hidratación agresiva y Ácido Zolendrónico 4 mg IV dosis única con resolución de su hipercalcemia y lesión renal aguda. Con dicho diagnóstico inició seguimiento por parte del servicio de Oncología con tratamiento a base quimioterapia tipo CHOP y con mejoría de su sintomatología.
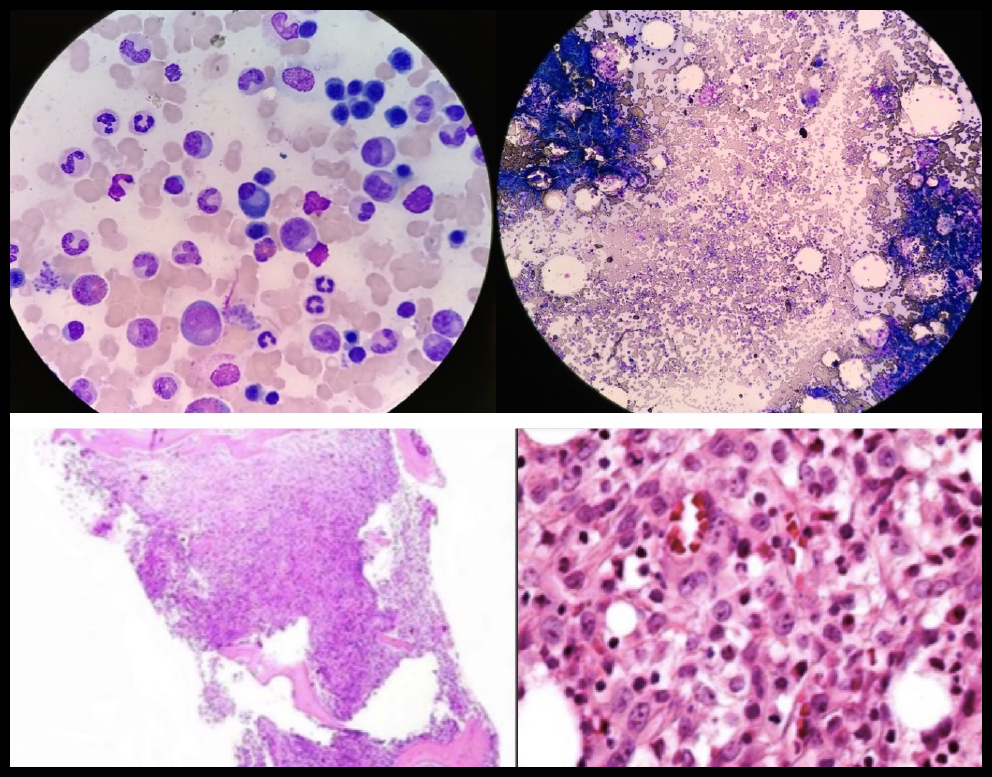
Figura 2. Biopsia y Aspirado de Médula Ósea: Infiltración de céulas malignas, 8% de células plasmáticas de distribución perivascular e intersticial, observandose médula ósea fibrosa con céulas grandes de abundante citoplasma, núcleos grandes y nucléolo prominente.
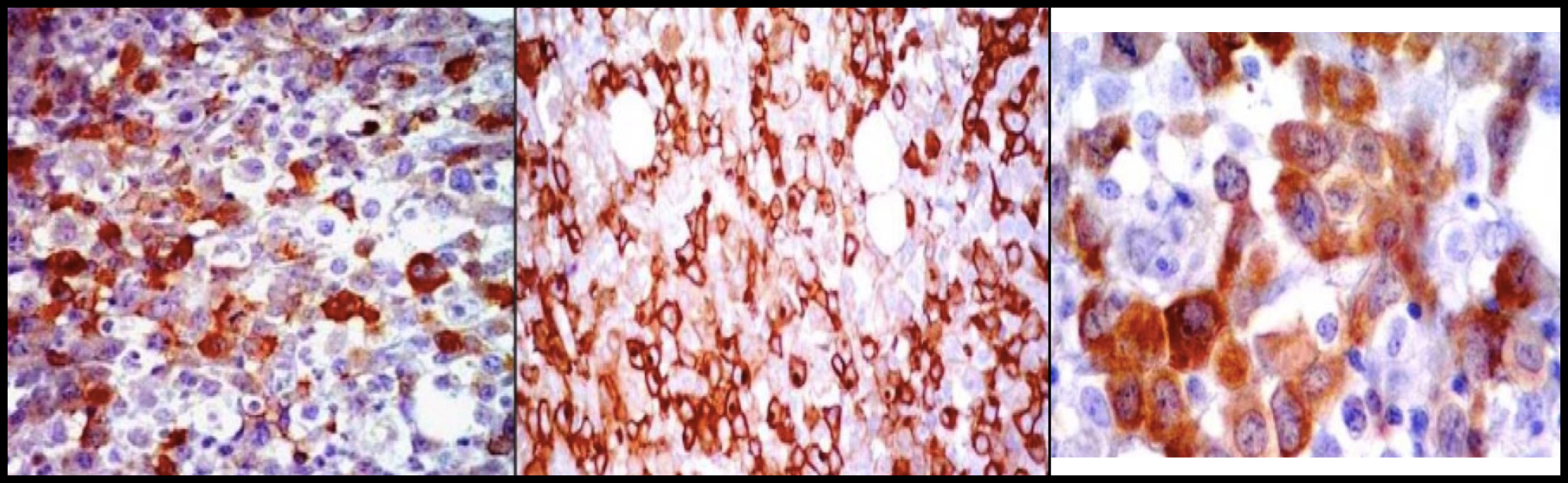
Figura 3. Inmunohistoquímica: Positividad para los marcadores del linfoma anaplásico EMA y CD30 con expresión en membrana citoplásmica y punto paranuclear, así como ALK-1 en núcleo y citoplasma.
DISCUSIÓN Y CONCLUSIÓN
La hipercalcemia en el contexto del linfoma de células T puede ser el resultado de varios mecanismos. La hipercalcemia asociada a un proceso maligno suele clasificarse como mediada por calcitriol, inducida por PTHrP e hipercalcemia osteolítica local. En nuestro paciente, parece que la principal causa de hipercalcemia fue el aumento de la producción de calcitriol por las células de linfoma maligno. Los linfomas de células T raramente se presentan con hipercalcemia, lesión renal aguda y lesiones óseas líticas, que suelen ser caraterísticas para el mieloma múltiple, como parte del CRAB. La electroforesis negativa de proteínas en suero y en orina, lesiones óseas líticas, anemia y lesión renal debe hacernos considerar diagnósticos alternativos como el linfoma. La diferenciación entre estas dos condiciones no puede hacerse sobre la base de imágenes o estudios generales de laboratorio. La biopsia de médula ósea y el muestreo de ganglios linfáticos son siempre necesarios para realizar el diagnóstico final.
REFERENCIAS
- Gerecke C, Fuhrmann S, Strifler S, Schmidt-Hieber M, Einsele H, Knop S. The diagnosis and treatment of multiple myeloma. Dtsch Arztebl Int 2016; 113 (27-28): 470–476.
- Landgren O, Kyle RA, Pfeiffer RM, Katzmann JA, Caporaso NE, Hayes RB, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood 2009; 113 (22): 5412–17.
- Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014; 15 (12): 538–48.
- Palumbo A, Anderson K. Multiple myeloma. N Engl J Med 2011; 364 (11): 1046–60.
- Lones MA, Sanger W, Perkins SL, Madeiros J. Anaplastic large cell lymphoma arising in bone. Report of a case of the monomorphic variant with the t(2;5) (p23;q35) translocation. Arch Pathol Lab Med 2000; 124 (9): 1339-43.
- Medeiros LJ, Elenitoba J. Anaplastic large cell lymphoma. Am J Clin Pathol 2007; 127 (5): 707-22.
- Delsol G, Ralfkiaer E, Stein H, Wright D, Jaffe ES. Anaplastic large cell lymphoma. In: World Health Organization Classification of Tumours. Pathology and genetics of tumours of haematopoietic and lymphoid tissues. IARC Press 2001; 94 (7): 230-5.
- Forteza J, Fraga M. Differential Diagnosis of Classic Hodgkin Lymphoma. Int J Surg Pathol Sup 2010; 18 (3): 124–127.
- Chor C. Shiu MA, Chi L. Malignant Lymphoma Masquerading as Multiple Myeloma. Leuk Lymphoma 1998; 28(5-6): 607-611.
- Chen H, Guimin Z, Lianjing W, Huichao Z, Xiaolin W. Simultaneous occurrence of Hodgkin’s lymphoma and multiple myeloma: A case report and review of the literature. Oncol lett 2016; 11 (6): 4139-4143.
INFORMACIÓN DEL ARTÍCULO
Autor para la correspondencia: Juan Daniel Díaz García. Dirección: Calzada de Tlalpan 4800, Tlalpan Centro I, Belisario Domínguez Secc 16, 14080 Tlalpan, Ciudad de México. Correo electrónico: judan_digar@hotmail.com. Télefono: +52 5534133330





